The Research Reactor TRIGA Mainz ŌĆō A Versatile Neutron Source for Basic Research, Applied Science and Education
K. Eberhardt and A. Kronenberg
Institut f├╝r Kernchemie, Universit├żt Mainz, D-55099 Mainz, Germany
Abstract
Currently, four research reactors with a thermal power ranging from 0.1 to 23 MWth are in operation in Germany and one new reactor (20 MWth) is under construction. The TRIGA Mark II reactor at the Institut f├╝r Kernchemie became first critical on August 3, 1965. It can be operated in the steady state mode with a maximum power of 100 kWth and in the pulse mode with a peak power of 250 MWth. A survey of the research programmes carried out at the TRIGA Mainz is given covering a wide range of applications in basic and applied science in nuclear chemistry, nuclear- and particle physics. Furthermore, the reactor is used for neutron activation analysis and for education and training of students and technical personal.
Momentan sind in Deutschland vier Forschungsreaktoren mit einer thermischen Leistung von 0,1 bis 23 MWth in Betrieb, ein neuer Reaktor (20 MWth) wird zur Zeit gebaut. Der Forschungsreaktor TRIGA Mainz des Instituts für Kernchemie wurde am 3. August 1965 erstmals kritisch. Er kann sowohl im Dauerbetrieb bei einer maximalen Leistung von 100 kWth, als auch im Pulsbetrieb mit einer Puls-Spitzenleistung von bis zu 250 MWth betrieben werden. Es wird ein Überblick über die verschiedenen Anwendungen in der Grundlagen- und angewandten Forschung in den Bereichen Kernchemie, Kern- und Teilchenphysik gegeben. Weiterhin wird der Reaktor für Neutronenaktivierungsanalysen, sowie in der Aus- und Weiterbildung von Studenten und technischem Personal genutzt.
1. Introduction
At the moment three MTR research reactors with a thermal power of 5 to 23 MWth and a TRIGA[1] reactor (100 kWth) are in operation in Germany. Furthermore, a research reactor with 20 MWth is under construction at the Technical University of Munich. TRIGA reactors are the most widely used research reactors in the world with steady state power levels ranging from 20 kWth to 16 MWth. The research reactors of the TRIGA type [1] are light water cooled reactors using fuel-moderator elements composed of an alloy of uranium-zirconium-hydride (UZrH) with 20% enrichment in 235U [2,3]. The fuel material in each element is in the form of a circular cylinder 35.5 cm long and approx. 3.5 cm in diameter. Graphite slugs at each end of the cylinder act as top and bottom reflectors. The elements are cladded either with aluminum or stainless steel.
The TRIGA reactorŌĆÖs inherent safety is due to a physical property of its fuel elements which gives the TRIGA core a large prompt negative temperature coefficient. Power rises initated by the rapid insertion of even the total available excess reactivity are automatically suppressed, and the reactor immediately returns to normal operating levels. In this reactor type the ŌĆ£warm neutrons principleŌĆØ is used. Much of its moderation of neutrons is due to the hydrogen that is mixed in with the fuel itself. Therefore, as the fuel temperature increases when a control rod (pulse rod) is suddenly removed by pressure, the neutrons inside the hydrogen-containing fuel rod become warmer than the neutrons outside in the cold water. These warmer neutrons inside the fuel cause less fissioning than the neutrons outside in the water. The end result is that the reactor automatically reduces power within a few thousandths of a second, faster than any engineered device can operate.
At the TRIGA Mainz, the reactor core is placed inside an aluminum tank of 2.00 m in diameter and 6.25 m in height, filled with distilled water. The shielding water above the core (4.9 m height) permit the easy, safe insertion and removal of samples during full-power operation. The fuel-moderator elements are fixed in the core with a top and bottom grid plate containing 91 positions. Positions not loaded with fuel-moderator elements, control-rod guide tubes or irradiation channels are filled with graphite dummy elements. Currently, the reactor core is equipped with 74 fuel elements, each containing about 36 g of 235U. A ring-shaped graphite reflector with a radial thickness of 30.5 cm surrounds the core. Figure 1 shows a vertical cross section view of the TRIGA Mainz indicating the core-reflector configuration and the position of some of the irradiation facilities.
In the steady state mode the reactor can be operated at power levels ranging from about 100 mWth up to 100 kWth, depending on the requirements of the different experiments. Pulse-mode operation is also possible [4,5]. The operation licence allows the insertion of an excess reactivity up to 2 dollar[2], corresponding to a pulse peak power of 250 MWth. Under these conditions, the pulse width (FWHM) is about 30 ms.
The TRIGA Mainz became first critical on August 3, 1965. In 1995, a complete refurbishment of the cooling and purification circuits took place and the cooling tower was renewed. The average operation time of the TRIGA Mainz is about 200 days per year. In the last five years approx. 90 % of this time was used for reactor operation at the nominal power of 100 kWth and the rest for pulses.
2. Irradiation Facilities and Pulse Mode Operation
2.1 Irradiation facilities
The TRIGA Mainz is equipped with four beam tubes. Figure 2 shows a horizontal section view of the TRIGA Mainz indicating arrangement and typical use of the four beam tubes. Two beam tubes are used for chemical experiments, mainly for the development of fast chemical separation procedures, continuous and discontinuous ones. These procedures are applied for the investigation of short-lived fission products and for studies of the chemical behaviour of the heaviest elements [6]. For a continuous transport of fission product activity from the production site near the reactor core to a detection unit or a separation facility, a gas-jet transport system [7] is used. For this, a target arrangement as shown schematically in figure 3 is applied. The fission targets consist of 235U, 239Pu or 249Cf, electrodeposited on a titanium or aluminium backing. Target thicknesses range from about 50 ┬Ąg/cm2 (249Cf) up to a few mg/cm2 (235U). Fission fragments leaving the target are thermalized in helium, argon or nitrogen as a carrier gas loaded with aerosol particles, e.g. metal halides or molybdenumoxide. The aerosol is produced in a furnace kept at a temperature of about 50 ┬░C below the melting point of the chosen material. In order to select particles with a size between 0.1 and 1.0 ┬Ąm a polyethylene tube of 6 mm diameter and a glass-frit are placed behind the aerosol generator. Attached to the surface of the aerosol particles the activity is transported to a device for chemical or physical separation by means of an outlet tube made of, e.g. polyethylene with an inner diameter of 1-2 mm (see fig. 3). With typical flow rates of 1-2 l/min., transport times in the gas-jet of about 1 s are possible. At the third beam port a neutron spin filter (NSF) for the production of polarized neutrons by interaction with optically pumped 3He atoms is installed [8]. The fourth beam tube will be used to create an intense beam of ultracold neutrons (UCN) by applying a very low temperature converter kept at a temperature of only a few degrees Kelvin or below [9].
For isotope production and for neutron activation analysis (NAA) the rotary specimen rack with 80 irradiation positions and the central thimble as indicated in figure 1 are most often used. Here, the samples are transferred manually. For the production of nuclides with half-lifes in the order of a few minutes, the TRIGA Mainz is equipped with three pneumatic transfer systems (so called ŌĆ£rabbitŌĆØ systems, see fig. 1). From a terminal located in the reactor hall or in a radiochemical laboratory a sample is transferred pneumatically to the irradiation position and back. Transport times of 1 to 5 seconds can be achieved. Neutron and g-irradiations of different biological and environmental samples, electronic equipment and for the calibration of neutron detectors are also performed at various irradiation positions, e.g., in a special sample holder close to the graphite reflector or on top of the thermal column. Table 1 compiles the neutron flux at the different irradiation positions in the steady state mode of 100 kWth.
2.2. Pulse mode operation
For pulse mode operation, the reactor is brought to criticality at a low steady state power, normally 50 W, and then a control rod is shot out of the reactor core with compressed air. Due to this sudden insertion of excess reactivity the power rises sharply with a reactor period of only a few milliseconds. The pulses have a shape that can be approximated by a Gaussian function with a width at half maximum in the millisecond range. By irradiating N atoms of an isotope having the neutron cross section s [10-24 cm2] and the half-life T1/2 [s] for the time t [s] with a neutron flux F [n cm-2 s-1] results in the acivity A [s-1] as expressed in general terms by equation (1). The decay constant l is defined as l = ln2/T1/2:
A = s├ŚF ├Ś N ├Ś (1 ŌĆō e-lt) (1)
The ratio of the pulse-generated activity Ap to the saturation activity AS, which is rapidly reached for short-lived nuclides under steady state conditions, is given by equation (2) - see [5] for a detailed discussion:
AP/AS = (0.737├Ś tP├Ś RP)/T1/2 (2)
Here, tP is the pulse width at half maximum and RP is the ratio of the pulse peak power and the maximum steady state power output (100 kWth). For a pulse peak power of 250 MWth, tP is ~30 ms. Introducing these data into (2) one obtains for the activity ratio AP/AS = 55 s/T1/2. Thus, for a 55 s-nuclide the activity produced with a 250 MWth pulse is equal to the saturation activity obtained by steady state irradiation. For a 70 MWth pulse the ratio is AP/AS = 38 s/T1/2 [5]. From this, one can estimate for which short-lived nuclides activation by pulsed irradiation is advantageous.
3. Research Programmes
3.1 Chemical on-line separation procedures
For the separation and subsequent detection of short-lived nuclides from complex reaction product mixtures as, e.g., obtained in thermal neutron-induced fission of 235U, 239Pu, and 249Cf or in heavy ion reactions, fast chemical procedures are required. Such procedures can be developed and optimized at the TRIGA Mainz by combining a gas-jet transport system with chemical separation facilities. Short-lived fission products with half-lifes down to 0.8 s have been investigated at the TRIGA Mainz [10-12] by means of the chemical separation system SISAK-3 (Short Lived Isotopes Studied by theAKUFE Technique). The SISAK method is based on fast multistage solvent extractions with specially designed high-speed centrifuges (15.000 - 30.000 rpm; 0.3 ml inner volume) for phase separation [13,14]. Figure 4 shows a typical SISAK set-up coupled to a gas-jet system installed at one of the TRIGA beam ports. In a degasser centrifuge the nuclear reaction products, as delivered from the gas-jet, are dissolved in an aqueous solution and the transport gas is removed. In a subsequent centrifuge, the aqueous phase is contacted with an organic solution containing an extractant for a selective and almost quantitative extraction of the desired element. If necessary, additional purification steps can be applied. After separation, the organic phase is pumped through a detector cell. Flow rates between 0.3 and 3 ml/s can be adjusted to optimize the hold-up time in the detector with respect to the half-life of the nuclide to be investigated.
Measurements of hold-up times in the different stages of the separation procedure can be performed by operating the TRIGA reactor in the pulse mode. An example of such a measurement is shown in figure 5 for the separation of technetium from the fission products of 249Cf. The aqueous phase (0.1 M H2SO4/0.01 M KBrO3) containing the activity is contacted with chloroform (CHCl3) as organic solvent, in which 10-4 M F4AsCl (tetraphenylarsoniumchloride) are dissolved as a selective extractant for Tc. In figure 5, the Tc-activity in the measuring cell at different flow rates (2.3, 1.3 and 0.9 ml/s, respectively) of the organic phase is plotted as a function of time after the reactor pulse. It serves as a start signal for the hold-up time measurement. The first peak is related to the burst of g-rays produced during the pulse. At a flow rate of 2.3 ml/s it takes about 3 seconds until the activity reaches the detector. This system has been applied to study the decay properties of the neutron rich isotopes 106-111Tc [10] and to determine level lifetimes in 106,108,110Ru [11] and 109,111,113Rh [12] by b-g/g coincidence measurements.
In chemical investigations of the heaviest (transactinide-) elements their chemical properties are compared with those of their lighter homologues. For the development of suitable separation procedures for the different transactinide elements, their lighter homologues in the periodic table of the elements are used. These are obtained by thermal neutron-induced fission of 235U, 239Pu or 249Cf. Such separation procedures in the gas phase and in aqueous solutions have been worked out, among others with fission nuclides of Zr, Nb, Mo, for chemical studies of Rf (rutherfordium, element 104), Db (dubnium, element 105), Sg (seaborgium, element 106) and, very recently, Bh (bohrium, element 107) [15].
3.2. 3He Neutron spin filter
A facility for the production of polarized neutrons by interaction with polarized 3He (
![]() ) has been build-up
and tested at the TRIGA Mainz. It consists of a neutron collimator installed in beam port B, a glass-cell placed between two Helmholtz-coils and filled with
) has been build-up
and tested at the TRIGA Mainz. It consists of a neutron collimator installed in beam port B, a glass-cell placed between two Helmholtz-coils and filled with
![]() under a pressure of 3 bar [8], a CoFe-crystal for neutron polarization analysis and a neutron counting device to determine the yield of polarized neutrons. Figure 6
shows a cross-section view of the neutron collimator. At the focus of the collimator the spin-filter cell (60 mm in diameter) is placed and exposed to a thermal neutron flux of 1 x 106 n*cm-2*s-1. The cell is coated with
Cs in order to provide relaxation times of the polarized 3He up to a 120 h. In the cell the reaction
under a pressure of 3 bar [8], a CoFe-crystal for neutron polarization analysis and a neutron counting device to determine the yield of polarized neutrons. Figure 6
shows a cross-section view of the neutron collimator. At the focus of the collimator the spin-filter cell (60 mm in diameter) is placed and exposed to a thermal neutron flux of 1 x 106 n*cm-2*s-1. The cell is coated with
Cs in order to provide relaxation times of the polarized 3He up to a 120 h. In the cell the reaction
![]() (n,p)T takes place.
(n,p)T takes place.
![]() has a high cross section for neutrons with antiparallel spin, whereas it is almost zero in the
case of parallel spin of the incoming neutron. In this way, the cell acts as a filter for neutrons with the unwanted spin component. Polarized neutrons with a yield of 84% at a total transmission of 12% for a neutron
wavelength of 1 ├ģ could be obtained with this arrangement [8]. This very effective neutron spin filter (NSF) has been investigated in a collaboration of the Institut f├╝r Physik, and the Institut f├╝r Kernchemie in Mainz, the
├ēcole Nationale Sup├®rieur in Paris, the Hahn-Meitner-Institut in Berlin, and the Institut Laue-Langevin (ILL) in Grenoble. Recently, such a NSF-device has been installed at the high flux reactor of the ILL in Grenoble in
order to perform neutron physics and neutron scattering experiments.
has a high cross section for neutrons with antiparallel spin, whereas it is almost zero in the
case of parallel spin of the incoming neutron. In this way, the cell acts as a filter for neutrons with the unwanted spin component. Polarized neutrons with a yield of 84% at a total transmission of 12% for a neutron
wavelength of 1 ├ģ could be obtained with this arrangement [8]. This very effective neutron spin filter (NSF) has been investigated in a collaboration of the Institut f├╝r Physik, and the Institut f├╝r Kernchemie in Mainz, the
├ēcole Nationale Sup├®rieur in Paris, the Hahn-Meitner-Institut in Berlin, and the Institut Laue-Langevin (ILL) in Grenoble. Recently, such a NSF-device has been installed at the high flux reactor of the ILL in Grenoble in
order to perform neutron physics and neutron scattering experiments.
3.3. Ultracold neutrons
Ultracold neutrons (UCN) are neutrons with a very low kinetic energy and wavelengths around 1000 ├ģ. They are totally reflected from the surface of most materials under any angle of incidence. This offers the possibility to guide them from the production site inside a reactor tank towards a storage facility like a so called neutron bottle or a magnetic trap for further investigations. It is planned to apply the pulse mode operation of the TRIGA Mainz for the production of high densities of UCN [16]. For this, a cryostat including a very low temperature converter consisting of solid deuterium or isotopically pure liquid 4He will be installed in beam tube C. A high quality neutron guide will focus the UCN beam to a storage facility for further experiments. The pulse mode operation of the TRIGA is advantegeous, since the neutron density during a pulse is in the order of 1015 n/cm2. The maximum pulse frequency is 12 pulses per hour at a pulse length of 30-40 ms. There is only a weak g-ray field resulting from such a reactor operation that will heat up the low temperature UCN source. UCN offer the possibility to study fundamental characteristics like the neutron lifetime and b-decay asymmetries or to measure the neutron electric dipole moment with great precision. The very low energy and momentum resolution that can be achieved with UCN opens the way for new applications in high resolution spectroscopy and scattering experiments including the investigation of biologicalmolecules [9].
4. Neutron activation analysis
Neutron activation analysis in combination with high-resolution gamma ray spectrometry is a versatile method for various analytical problems due to its simplicity, multielement capacity, and sensitivity. The instrumental neutron activation analysis (INAA) is performed without any chemical separation steps, whereas the radiochemical neutron activation analysis (RNAA) applies chemical procedures either prior to or after the neutron irradiations, mostly in a research reactor. Figure 8 show the detection limit achieved by INAA with the TRIGA Mainz. The delayed neutron activation analysis (DNAA) is a special version and uses the counting of b-delayed neutrons emitted from very neutron-rich fission products.
In principle, all elements with Z ┬│ 9 (with few exceptions) can be analyzed without any influence of the matrices or the chemical bonding [17, 18]. The modern high- resolution g-spectrometry can simultaneously determine up to 30-40 elements, with a sensitivity in the range of ppb (ng/g). In special cases the selectivity and the detection limit of the neutron activation analysis can be improved by applying coincidence or anticoincidence systems reducing the Compton background [22-24].
Neutron activation analysis can be performed in two different ways, either as an indirect method relative to standard samples, or as an absolute technique using tabulated reactor-specific k0-values [25]. Special and temporary fluctuations of the neutron flux are monitored using 197Au.
Neutron activation analysis was and is still widely used in various research fields, such as archaeology, biology, environmental and food monitoring, forensic science as well as in material science and industry [17-21]. This technique is also employed for the certification of standards, and for the validation of other analytical methods.
4.1 Features of the Delayed Neutron Activation Analysis (DNAA)
Delayed neutron activation analysis is used for the fast determination of fissile nuclides, such as 233U, 235U,239Pu, 232Th etc. by measuring the delayed neutron emission after fission [26].
From the nuclides produced in nuclear fission, about 110 are known precursors of b- delayed neutrons emission with b-half-lives ranging from milliseconds to minutes [27]. Delayed neutrons emitted by the long-lived isotopes 88Br, 137I, and 87Br are generally measured. The sensitivity is less than 10-11 g for 235U, 10-6 g for 232Th and 10ŌĆæ11 g for 239Pu under conditions of the TRIGA Mainz.
For the determination of thorium in samples containing uranium, a cadmium cover for the absorption of thermal neutrons is used eliminating delayed neutrons from uranium fission. Thorium fissions only with epithermal neutrons. To increase the sensitivity for thorium a chemical separation can be applied.
Typically, the samples are irradiated for 2 min in the reactor and after a delay time of 15 to 20 s, they are counted for 1 min with 3He proportional tubes in a circular arrangement. The delayed neutrons thermalized by paraffine and polyethylene are detected with an efficiency of 33%.
This method is routinely used for the incorporation inspection of workers from the uranium industry. Generally a-spectroscopy is employed for such investigations, but DNAA is more simple and faster. Knowing the isotopic composition of uranium a screening of a large number of persons can be carried out with the precision demanded by the authorities.
4.2 Examples for the application of neutron activation analysis
In cosmo- and geochemistry, neutron activation analysis is used together with spark mass spectrometry for trace analysis of extraterrestrial samples [28]. Among others, samples from the Moon as obtained from the Apollo 11 expedition [29], the Mars meteorite ALH 84001 [30] and a Mars meteorite found in 1998 in the Libyan desert [31] were analyzed by INAA.
Numerous geological samples have been measured in the last few years. Important contributions came from the analysis of highly siderophile elements (noble metals, Re, and Au) by the nickel sulfide technique in combination with neutron activation analysis [32]. The element pattern and ratios give important information about extraterrestrial components in melts from meteoritical impacts.
For environmental monitoringRNAA has very often been used. Examples are the determinations of platinum-group elements (PGE) in soil samples nearby a highways in connection with catalytic converters of cars [33], and the determination of AOX (Absorbable Organic Halogens) [34]. Sediments from a former Ag-mine in Freiberg/Saxonia were investigated with INAA [35] yielding a relative high content of arsenic, rare earth elements, thorium, and uranium. Investigations on samples from former Hg-mine of the Nordpf├żlzer Bergland [36] and from the uranium mining in former East Germany (Wismut) were also performed [37]. For the Wismut samples, uranium from water and river sediments (ŌĆ£Wei├¤e ElsterŌĆØ) were analysed by INAA and DNAA in order to study the transport processes between slagheap to the river [37].
INAA in combination with anti-compton spectrometry has been used for air filter analysis. The high detection efficiency and the high reliability of the detection system allowed detailed monitoring of 36 elements simultaneously. With the fingerprint of these elements, it was possible to distinguish between natural and anthropogenic origin [38].
5. Isotope production, neutron- and g-irradiations
Radioactive isotopes are widely used in research and industry, e.g. for calibration of radiation detectors, to trace chemical reactions, for density and thickness measurements or for migration studies. Various radioactive isotopes are produced at the TRIGA Mainz. Among them are 24Na, 32P, 41Ar, 71Ge, 76As and 239Np. For monitoring liquid and gaseous flow patterns and to measure hold-up times in chemical reactors installed in industrial production plants 24Na (in form of its acetate) and 41Ar (chemically bound in hydrochinone) are used. The b-emitting isotope 32P is used for calibration purposes in b-spectroscopy. 71Ge and 76As serve as yield tracers to develop and synthesize new radiopharmaceuticals. For studies of the chemical behaviour of carrier-free amounts of neptunium under environmental conditions the 2.3 d - 239Np can be produced via the reaction 238U(n,g)239U and subsequent b-decay into the 239Np.
Neutron and g-irradiations of samples with a volume up to a few litres can be performed in special containers made of aluminum or polyethylene placed near the reactor core, e.g. on top of the thermal column (see fig. 1). Typical examples are studies of radiation damage in electronic components employed in high energy physics particle detectors, the effect of high g-dose rates on the destruction of plant seeds or the application of g-irradiation as a non-chemical disinfectant in microbiology. A distinct position in the reactor pool is used for the calibration of neutron- and g-probes to be installed in nuclear power plants.
6. Education
As part of the department of chemistry at the University of Mainz various courses in nuclear and radiochemistry, radiation protection and reactor physics are held at the Institut f├╝r Kernchemie for advanced students, teachers, engineers and technicians. In the following an overview on the use of the TRIGA reactor within the frame of this educational programmes is given:
┬Ę In the basic course in nuclear chemistry neutron activation analyses are performed to demonstrate the capabilities of this method for trace analysis of various materials. Furthermore, the level-scheme of the 5.3 min - 190mOs is investigated by means of g-spectroscopy after irradiations of Os-samples in the rabbit system. After irradiation of isotopically enriched 235U with thermal neutrons, Ba is separated radiochemically to demonstrate nuclear fission. Experiments to investigate the chemistry of neptunium as the first transuranium element are also performed.
┬Ę As part of the experimental programme of the course in radiation protection, measurements of the neutron- and g-dose rates at the biological shield and near the surface of the reactor pool water are performed. Here, the reactor is operated at different power levels from a few Watt to 100 kWth and the dose rates are monitored as a function of reactor power. Similar dose rate measurements at an open beam port are also performed.
┬Ę The course in reactor physics and reactor operation consists of a lecture in reactor physics and practical training in reactor operation at steady state power as well as in the pulse mode. Among other experiments, the effect of a strong neutron absorber on the reactivity is investigated by introducing various amounts of Cd (in form of bulk material and as Cd-acetate). The change of the position of the control rods for the different samples is registered. After a calibration of the control rods using the inhour-method the reactivity change can be deduced.
In addition to the above mentioned courses guided tours are carried out frequently by the staff of the TRIGA Mainz. 40-60 groups, corresponding to 600-800 people, visit the institute every year.
7. Outlook
The TRIGA Mainz is one of only four research reactors that are currently in operation in Germany. It is intensively used for basic research, applied science and educational purposes and is in an excellent technical state. Recently it was decided that the reactor will be operated at least until the year 2010. This decision was mainly made in connection with the research work performed and planned at the TRIGA Mainz and the training of students in the fields of nuclear chemistry, nuclear physics and radiation protection. Taking into account the past and future operation schedule and the typically low burn-up of TRIGA fuel elements (~4g 235U/a), the reactor can be operated for the next decade with the fresh fuel elements on stock and without changing spent fuels.
References:
[1] Koutz, S.L.; Taylor T.; McReynolds, A.; Dyson, F.; Stone, R.S.; Sleeper, H.P.; Duffield, R.B.: Design of a 10 kW Reactor for Isotope Production, Research and Training Purposes. Proc. 2nd U.N. Intern. Conf. Peaceful Uses Of Atomic Energy (1958) 1017
[2] Merten, U.; Dijkstra, L.J.; Carpenter, F.D.; Hatcher, A.P.; La Grange, L.D.: The Preparation and Properties of Zirconium-Uranium-Hydrogen Alloys. Proc. 2nd U.N. Intern. Conf. Peaceful Uses Of Atomic Energy (1958) 789
[3] McReynolds, A.W.; Nelkin, M.S.; Rosenbluth, M.N.; Whittemore, W.L.: Neutron Thermalization by Chemically-bound Hydrogen and Carbon. Proc. 2nd U.N. Intern. Conf. Peaceful Uses Of Atomic Energy (1958) 1540
[4] Stone, R.S.; Sleeper, H.P.; Stahl, R.A.; West, G.: Transient Behaviour of TRIGA, a Zirconium-Hydride, Water - Moderated Reactor. Nuclear Science and Engineering 6 (1959) 255
[5] Menke, H., Trautmann, N.; Krebs, W.-J.: Irradiations by means of reactor pulses. Kerntechnik 17 (1975) 281
[6] Trautmann, N.: Fast Radiochemical Separations for Heavy Elements. Radiochim. Acta 70/71 (1995) 237
[7] Stender, E.; Herrmann, G.; Trautmann, N.: Use of Alkali Halide Clusters in a Gas-Jet Recoil Transport System. Radiochem. Radioanal. Letters 42 (1980) 291
[8] Surkau, R.; Becker, J.; Ebert, M.; Grossmann, T.; Heil, W.; Hofmann, D.; Humblot, H.; Leduc, M.; Otten, E.W.; Rohe, D.; Siemensberger, K.; Steiner, U.; Tasset, F.; Trautmann, N.: Realization of a broad band neutron spin filter with compressed, polarized He-3 gas. Nucl. Instr. Meth. Phys. Res. A384 (1997) 444
[9] Steyerl, A.: Ultracold Neutrons: Production and Experiments. Physica B 156/157 (1998) 528
[10] Altzitouglou, T.; Rogowski, J.; Sk├źlberg, M.; Alstad, J.; Herrmann, G.; Kaffrell, N.; Skarnemark, G.; Talbert, W.; Trautmann, N.: Fast Chemical Separation of Technetium from Fission Products and Decay Studies of 109Tc and 110Tc. Radiochim. Acta 51 (1990) 145
[11] Schoedder, S.; Lhersonneau, G.; W├Čhr, A.; Skarnemark, G.; Alstad, J.; N├żhler, A.; Eberhardt, K.; ├äyst├Č, J.; Trautmann, N.; Kratz, K.-L.: Level Lifetimes in Neutron Rich Ru Isotopes. Z. Phys. A352 (1995) 237
[12] Lhersonneau, G.; Pfeiffer, B.; Alstad, J.; Dendooven, P.; Eberhardt, K.; Hankonen, S.; Kl├Čckl, I.; Kratz, K.-L.; Malmbeck, R.; Omtvedt, J.P.; Penttila, H.; Schoedder, S.; Skarnemark, G.; Trautmann, N.; Ayst├Č, J.: Spape Coexistence Near the Double-Midshell Nucleus 111Rh. Eur. Phys. J. A1 (1998) 285
[13] Persson, H.; Skarnemark, G.; Sk├źlberg, M.; Alstad, J.; Liljenzin, J.O.; Bauer, G.; Haberberger, F.; Kaffrell, N.; Rogowski, J.; Trautmann, N.: SISAK 3 ŌĆō An Improved System for Rapid Radiochemical Separations by Solvent Extraction. Radiochim. Acta 48 (1989) 177
[14] Alstad, J.; Skarnemark, G.; Haberberger, F.; Herrmann, G.; N├żhler, A.; Pense-Maskow, M.; Trautmann, N.: Development of New Centrifuges for Fast Solvent Extraction of Transactinide Elements. J. Radioanal. Nucl. Chem. 189 (1995) 133
[15] Kratz, J.V., Chemical Properties of the Transactinide Elements, p. 129-193, in Greiner, W. and Gupta, R.K. (ed.), Heavy Elements and Related New Phenomena, World Scientific, Singapore, New Jersey, London, HongKong, 1999
[16] Golub, R.: Ultracold Neutrons (UCN) at a TRIGA Reactor. Nucl. Instr. Meth. Phys. Res. 226 (1984) 558
[17]Desoete, D., Gigbels, R., Hoste, J.: Neutron Activation Analysis, Wiley-Interscience, London 1972
[18] Pfrepper, G., G├Črner, W., Niese, S., Spurenelementbestimmung durch Neutronenaktivierung, Akademische Verlagsgesellschaft Geest&Portig, Leipzig 1981
[19] Holzhey, J., Aktivierungsanalyse. Anwendung in der Metallurgie. VEB Dtsch. Vlg f. Grundstoffind., Leipzig 1975
[20] Laue, J.C., Neutron Activation Analysis of Geological Materials, At. Energy Rev. 17 (1979) 603-698
[21] Ehmann, W.D., Robertson, J.D., and Yates, S.W., Nuclear and Radiochemical Analysis, Anal. Chem. 64 (1992) 1R-22R
[22] Roesick, U., and Braetter, P., Z. Anal. Chem. 286 (1977) 336
[23] Mauerhofer, E. et al., A Compton suppression spectrometer for neutron activation analysis, Nucl. Instrum. Meth. A371 (1996) 465-471
[24] Mauerhofer, E., Improvement in the Counting Statistics and in the Limit of Detection with Compton Suppression Spectrometers ŌĆō a Contribution to Instrumental Neutron Activation Analysis, Appl. Radiat. Isot. 47 (1996) 649-658
[25] Simonits, A. L., Moens, F., De Corte, A., J. Radioanal. Chem. 60 (1980) 461
[26] Amiel, S., Anal. Chem. 34 (1962) 1683-1692
[27] Tomlinson, L., Delayed Neutron Precursors, Atomic Data and Nuclear Data Tables 12 (1973) 179-194
[28] W├żnke, H., Constitution of terrestrial planets, Phil. Trans. R. Soc. Lond. A303 (1981) 287-302 and H. W├żnke and G. Dreibus, Chemical composition and accretion history of terrestrial planets, Phil. Trans. R. Soc. Lond. A325 (1988) 545-557
[29] W├żnke, H., Chemistry of the Moon, Topic in Current Chemistry, Springer-Verlag, Berlin, Heidelberg, New York 1974
[30] Dreibus, G., et al., Chemical and Mineral Composition of ALH 84001: A Martian Orthopyroxenite, Meteoritcs & Planetary Science 29 (1996) 461
[31] Zipfel, J. et al. Petrology and chemistry of the new shergottite Dar al Gani 476, Meteoritcs & Planetary Science 35 (2000) 95-106
[32] Snow, J. E. and Schmidt, G., Constraints on Earth accretion deduced from noble metals in the oceanic mantle, Nature 391 (1998) 166
[33] Heinrich, E., Schmidt, G., Kratz, K.-L., Deterimation of platinum-group Elements (PGE) from catalytic converters in soil by means of docimasy and INAA, Fres. J. Anal. Chem. 354 (1996) 883-885
[34] Rollinger, D., Kratz, K.-L., Validation of neutron activation as analytical method for the determination of absorbable halogens in aqueous samples, J. Trace Microprobe Techn. 14 (1996) 255-264
[35] Schl├Čsser, D., Baacke, D., Beuge, P., Kratz, K.-L., Elemental compostion of sediments from former silver mine in Freiberg/East Germany, Appl. Rad. and Isot.50 (1999) 609-614
[36] Schl├Čsser, D., PhD Thesis, Institute for Nuclear Chemistry, University of Mainz, 1999
[37] Kupsch,H. (Projektleiter), Abschlu├¤bericht 1998 des Forschungsvorhabens des S├żchsischen Staatsministeriums f├╝r Umwelt und Landesentwicklung, ŌĆ×Radioaktive Belastung im Bereich von Altbergbauhalden ŌĆō Austrag radioaktiver Schadstoffe aus Halden des ErzbergbausŌĆ£; IIF-Leipzig, Leipzig, April 1998
[38] Nett, M., PhD Thesis, Institute for Nuclear Chemistry, University of Mainz, 1999
[39] Kratz, K.-L., 100 kW f├╝r den Umweltschutz, Forschungsmagazin der Johannes Gutenberg-Universit├żt Mainz 13 (1997) 86-94
|
Irradiation Position |
Thermal flux [n/cm2s]* |
Epithermal flux [n/cm2s]** |
|
Rotary specimen rack |
7 x 1011 |
4 x 1010 |
|
Rabbit systems |
1.6-1.8 x 1012 |
4.6-5.6 x 1010 |
|
Beam tubes |
1.0-5.4 x 1011 |
7.6 x 108 ŌĆō 1.6 x 1010 |
|
Central thimble |
4.2 x 1012 |
1.4 x 1011 |
|
Other positions |
107 ŌĆō 1010 |
102 ŌĆō 108 |
*EN ┬Ż 0.4 eV; **EN┬│ 0.4 eV
Table 1: Thermal and epithermal neutron fluxes of the different irradiation positions at the TRIGA Mainz for a power of 100 kWth.
Figures:
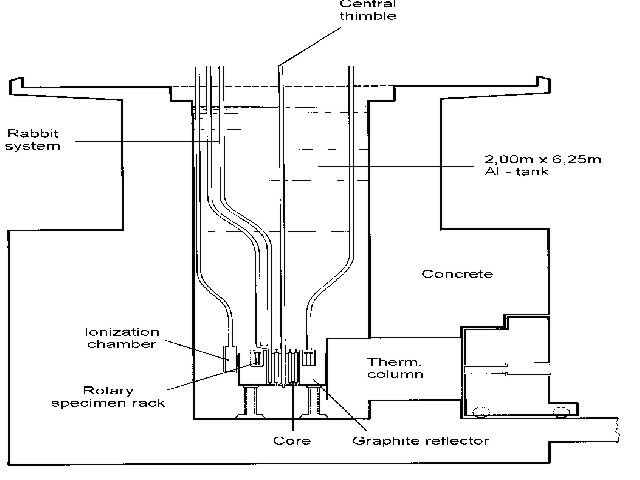
Fig. 1: Vertical cross section view of the TRIGA Mainz indicating the position of the reactor core surrounded by a 30.5 cm thick graphite reflector. For isotope production and neutron activation analysis the central thimble, the rotary specimen rack and the pneumatic transfer systems are mostly used.
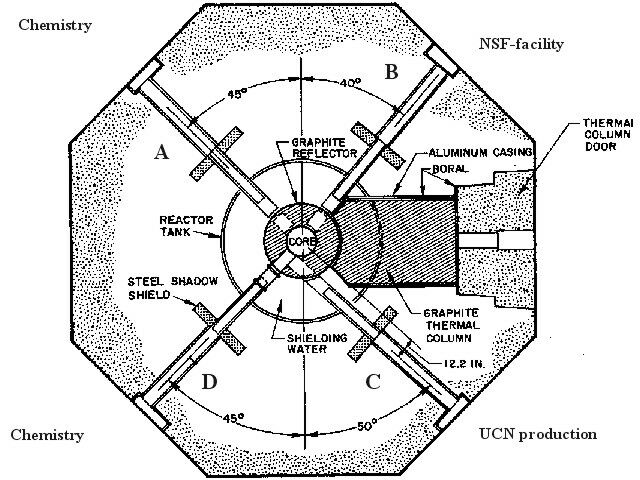
Fig. 2: Horizontal section view of the TRIGA Mainz. Beam ports A and D are mainly used for radiochemical investigations. Beam port B has been modified for the production of polarized neutrons by interaction with polarized 3He in a special NSF-cell. In port C the UCN-facility will be installed.
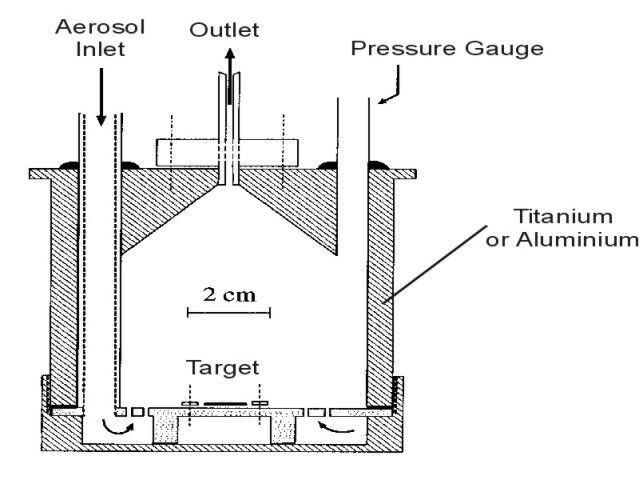
Fig. 3: Cross section view of a target arrangement used at the TRIGA Mainz. The target is fixed inside a cylindrical chamber made of titanium or aluminum. Fission fragments recoiling out of the target are thermalized in the carrier gas atmosphere and attached to the surface of aerosol particles.
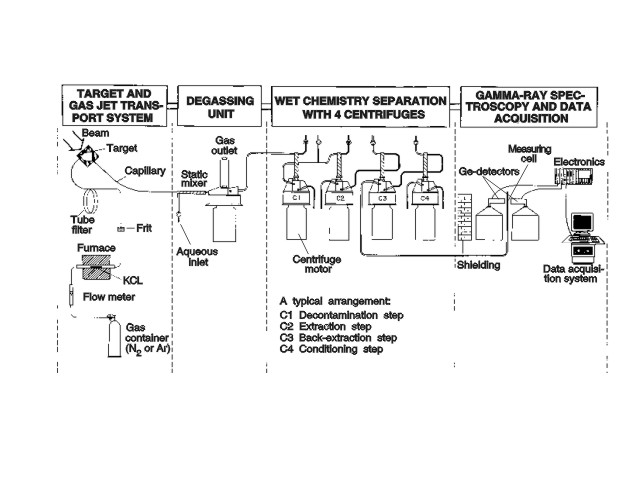
Fig. 4: Experimental set-up of the SISAK centrifuge system for multistage extraction
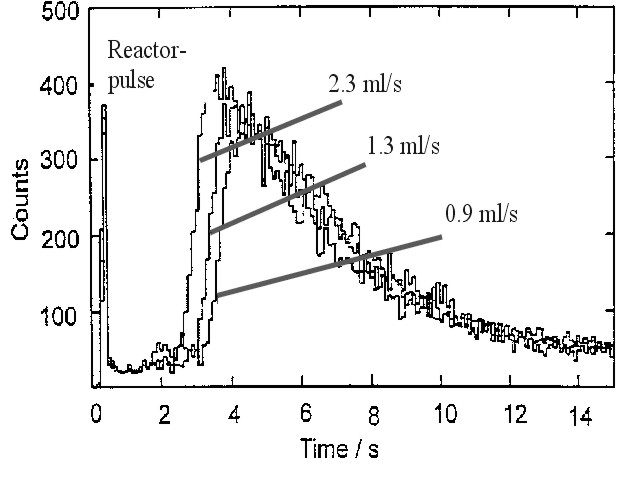
Fig. 5: Measurement of the hold-up time for a single-stage extraction of Tc produced in nuclear fission of249Cf. At a flow rate of 2.3 ml/s it takes approx. 3 seconds until the activity reaches the detector.
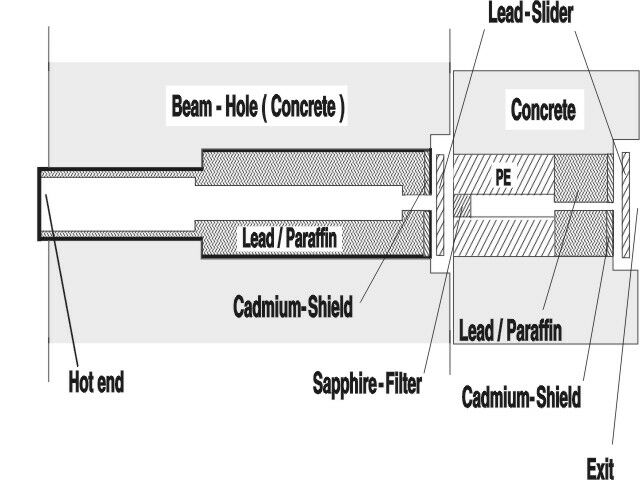
Fig. 6: Cross section view of the neutron collimator in beam port B. A spin filter cell filled with polarized 3He is placed near the exit and exposed to a neutron flux of 1x106 n*cm-2*s-1.
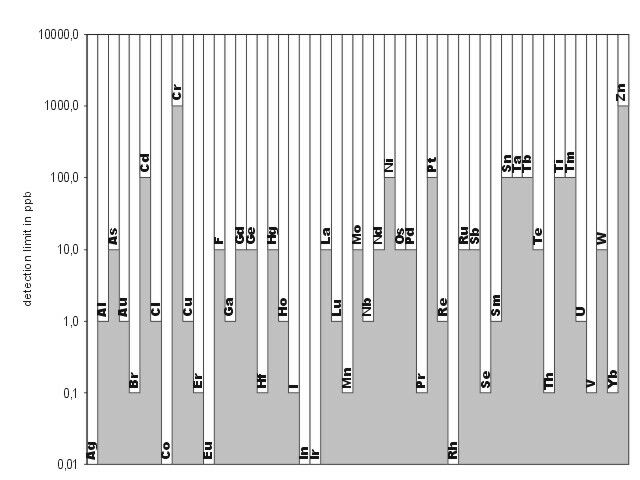
Fig. 7: Detection limit achieved by INAA at the TRIGA Mainz for a neutron flux of 1012 n*cm-2*s-1 for a number of elements (from [39])
[1] TRIGA is an acronym and means: Training Research Isotopes General Atomics
[2] The term dollar is used to denote the reactivity introduction that makes a reactor
prompt critical. For the TRIGA 1 dollar = 0.0073 [1].



Living textbook
of Nuclear Chemisty